seqlinkage
Construct phylogenetic tree from pairwise distances
Syntax
Description
Examples
Create an array of structures representing a multiple alignment of amino acids:
seqs = fastaread('pf00002.fa');Measure the Jukes-Cantor pairwise distances between sequences:
distances = seqpdist(seqs,'method','jukes-cantor','indels','pair');
Build the phylogenetic tree for the multiple sequence alignment from calculated pairwise distances. Specify the method to compute the distances of the new nodes to all other nodes. Provide leaf names:
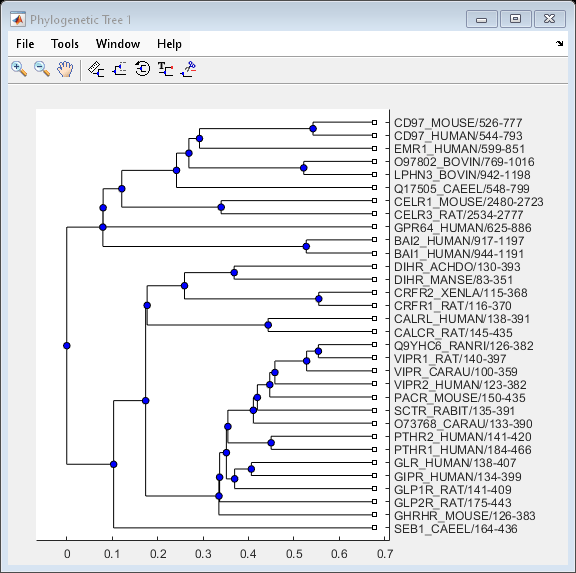
phylotree = seqlinkage(distances,'single',seqs)Phylogenetic tree object with 32 leaves (31 branches)
View the phylogenetic tree:
view(phylotree)
Input Arguments
Name-Value Arguments
Version History
Introduced before R2006aSee Also
phytree | phytreewrite | seqpdist | seqneighjoin | cluster | plot | view