cluster
Validate clusters in phylogenetic tree
Syntax
Description
LeafClusters = cluster(Tree,Threshold)
Starting with two clusters (k =
2), selects the partition that optimizes the criterion specified by theCriterionargument.Increments k by
1and again selects the optimal partitionContinues incrementing k and selecting the optimal partition until a criterion value =
Thresholdor k = the maximum number of clusters (that is, number of leaves)From all possible k values, selects the k value whose partition optimizes the criterion
[ returns a
column vector containing the cluster index for each leaf node and branch node in
LeafClusters,NodeClusters]
= cluster(Tree,Threshold)Tree.
[
returns a two-column matrix containing, for each step in the algorithm, the index of the
branch being considered and the value of the criterion. Each row corresponds to a step in
the algorithm. The first column contains branch indices, and the second column contains
criterion values.LeafClusters,NodeClusters,Branches] = cluster(Tree,Threshold)
___ = cluster(___,
) specifies options using one or more name-value arguments in addition to the input
arguments in previous syntaxes. For example, use Name=Value)MaxClust to specify the
maximum number of possible clusters for the tested partitions.
Examples
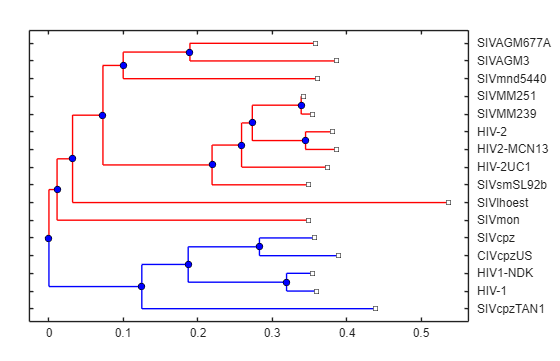
Read sequences from a multiple alignment file into a MATLAB structure.
gagaa = multialignread("aagag.aln");Build a phylogenetic tree from the sequences.
gag_tree = seqneighjoin(seqpdist(gagaa),equivar=gagaa);
Validate the clusters in the tree and find the best partition using the gain criterion.
[i,j] = cluster(gag_tree,[],Criterion="gain",Maxclust=10);Use the returned vector of indices to color the branches of each cluster in a plot of the tree.
h = plot(gag_tree); set(h.BranchLines(j==2),Color="b") set(h.BranchLines(j==1),Color="r")
Input Arguments
Name-Value Arguments
Output Arguments
References
[1] Dudoit, S. and Fridlyan, J. (2002). A prediction-based resampling method for estimating the number of clusters in a dataset. Genome Biology 3(7), research 0036.1–0036.21.
[2] Theodoridis, S. and Koutroumbas, K. (1999). Pattern Recognition (Academic Press), pp. 434–435.
[3] Kaufman, L. and Rousseeuw, P.J. (1990). Finding Groups in Data: An Introduction to Cluster Analysis (New York, Wiley).
[4] Calinski, R. and Harabasz, J. (1974). A dendrite method for cluster analysis. Commun Statistics 3, 1–27.
[5] Hartigan, J.A. (1985). Statistical theory in clustering. J Classification 2, 63–76.
Version History
Introduced before R2006a
See Also
Objects
Functions
phytreeread|phytreeviewer|seqlinkage|seqneighjoin|seqpdist|plot|view|cluster|silhouette